
 Descargar número completo
Descargar número completo Download full issue
Download full issueCITA ESTE TRABAJO
Fernandez Carrasco M, López González J, Campos Serrano N. Descompensación edemoascítica: ¿es siempre cirrosis hepática?. RAPD 2024;47(5):198-200. DOI: 10.37352/2024475.3
Caso clínico
Varón de 72 años sin antecedentes personales de interés ni hábitos tóxicos, que ingresa por diarrea de dos meses de evolución, asociada a síndrome constitucional, ascitis y edemas de nueva aparición.
Analíticamente destaca ligera leucocitosis con neutrofilia, leve deterioro de la función renal con creatinina 1.34mg/dl e hipoalbuminemia, con plaquetas, perfil hepatobiliar y coagulación normales.
Se realizan durante el ingreso ecografía abdominal, tomografía computerizada (TC) y angioTC (imágenes) donde se aprecian hallazgos sugerentes de hepatopatía crónica y destaca muy llamativa dilatación de la porta principal y ambas ramas intrahepáticas, con disminución de calibre a nivel de la vena cava inferior, en el segmento de entrada al hígado que aparece prácticamente filiforme, así como calibre reducido del segmento proximal de las venas suprahepáticas, siendo estas permeables, sin esplenomegalia asociada. Se realiza endoscopia digestiva alta que descarta la presencia de varices y ecocardiografía, sin alteraciones.
Durante el ingreso el paciente evoluciona de forma favorable, manteniendo buen ritmo de diuresis, disminución de edemas y de perímetro abdominal con tratamiento deplectivo, llegando al diagnóstico de HP posthepática secundario a obliteración membranosa de la vena cava inferior.
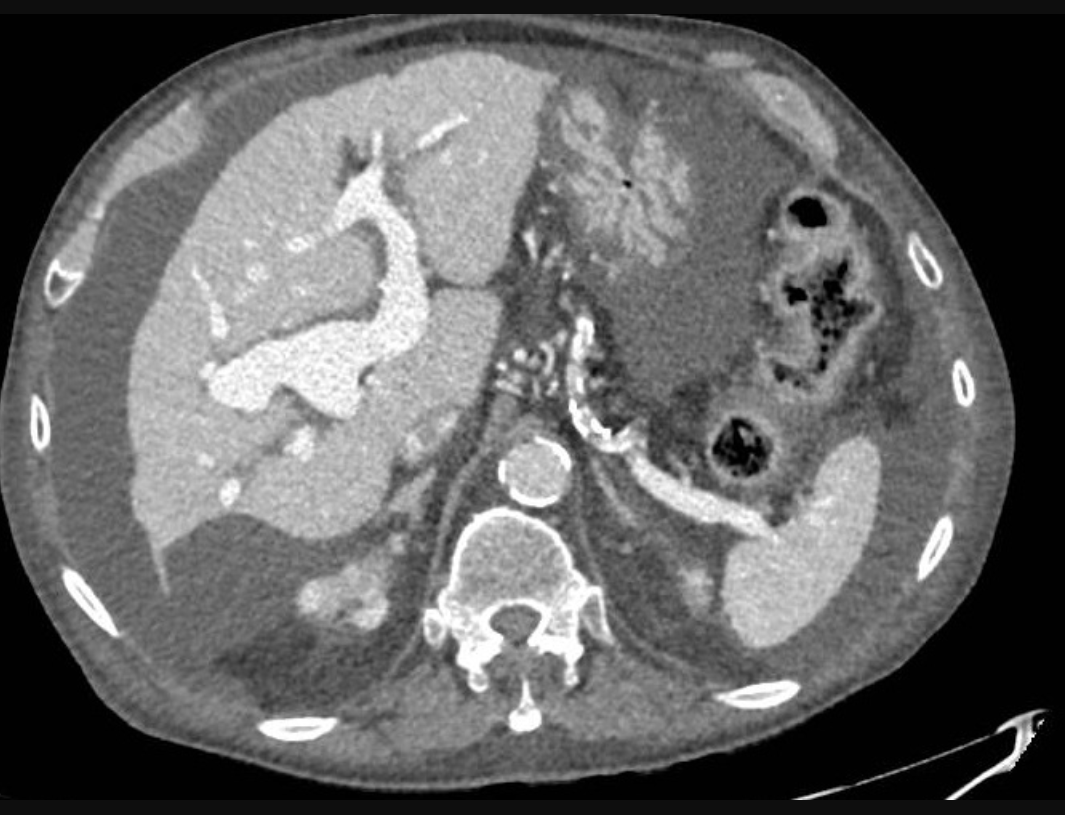
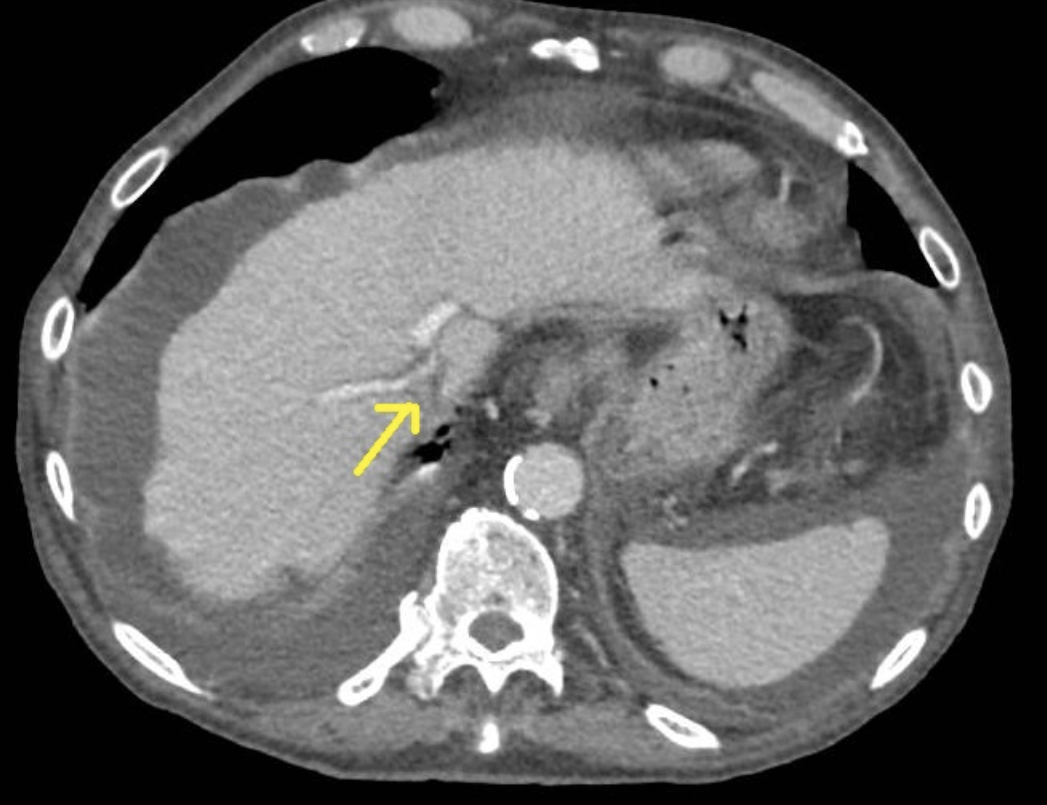
Discusión
La hipertensión portal posthepática se produce cuando existe un deterioro en el flujo venoso de salida hepático, aumentándose las resistencias a este nivel[1]. La causa más frecuente de HP posthepática es el síndrome de Budd-Chiari (SBC), la insuficiencia cardiaca derecha o la obstrucción de la vena cava inferior por tumores, trombosis o aumento de caudado[2].
El SBC es una entidad clínica que engloba un conjunto de síndromes que pueden provocar obstrucción del drenaje venoso hepático a cualquier nivel, desde las pequeñas venas hepáticas hasta la unión de la VCI con la aurícula derecha[2].
Es una entidad poco frecuente. Las principales causas son neoplásicas, hematológicas, inflamatorias, infecciosas y farmacológicas. Sin embargo, tras descartar estas etiologías se debe pensar en otras menos frecuentes como la oclusión membranosa de la VCI (OMVCI), conocida también como hepatocavopatía obliterativa, o síndrome de la vena cava hepática[3].
En cuanto a la fisiopatología se ha propuesto un origen congénito debido a un fallo en la embriogénesis de la vena cava inferior (VCI), aunque, en distintos estudios se establece el probable origen adquirido o la posible transformación membranosa que provoca una estenosis a nivel de la vena cava inferior[4].
Cínicamente se manifiesta con signos de HP. A nivel analítico no existe gran alteración de la función hepática. También se ha asociado al desarrollo de hepatocarcinoma, a priori menos agresivo que el debido a otras etiologías. Las pruebas de imagen nos darán el diagnóstico, siendo el signo del "flujo inverso de la VCI" y el signo del "sangrado en jet" en TC y RM específicos del SBC subtipo obliteración membranosa[5].
El diagnóstico se basa en características clínicas mencionadas anteriormente y la visualización en pruebas de imagen, pudiendo distinguir una oclusión completa de parcial. La tomografía computarizada o la cavografía mediante la administración de contraste, pueden resaltar el nivel de la obstrucción a partir del cual se produce una fuga retrógrada o signo del flujo inverso de la VCI[3].
El tratamiento de elección de la OMVCI es la recanalización de la zona obstruida de forma endovascular, mediante la angioplastia percutánea con balón, siendo eficaz hasta en un 91% de los casos. Existen otras técnicas quirúrgicas menos empleadas como la membranotomía, membranectomía o realización de shunts[1],[3]. Las complicaciones asociadas a la HP se tratan de forma similar al resto de etiologías.
Actualmente, la OMVCI se engloba entre las posibles causas del síndrome de Budd- Chiari, aunque algunos autores sugieren que se podría considerar un síndrome distinto[3]. La importancia radica en la posibilidad de un tratamiento curativo, el cual evitaría la elevada morbimortalidad secundaria a las descompensaciones por HP, así como el riesgo de desarrollar hepatocarcinoma.
