
 Descargar número completo
Descargar número completo Download full issue
Download full issueCITA ESTE TRABAJO
Rodríguez González D, Ruiz Ginés MÁ, Ruiz Ginés JA, Moreno Torres B, Lorenzo Lozano MC. Hepatopatía glucogénica: descripción de un caso atípico. RAPD Online. 2021;44(3):105-109.
Introducción
El inicio de la terapia insulínica en el tratamiento de la diabetes mellitus tipo 1 (DM1) permitió observar (Mauriac, 1930)[1], por primera vez, en pacientes mal controlados, la aparición de un síndrome caracterizado por talla baja, retraso puberal, rasgos cushingoides, hepatomegalia y elevación de enzimas hepáticas (síndrome de Mauriac)[1]. Posteriormente, las alteraciones hepáticas descritas en pacientes diabéticos han sido numerosas, predominando la elevación de enzimas hepáticas, hepatomegalia y depósito hepático intracelular de glucógeno. Se han descrito casos sindrómicos incompletos, tanto en niños como en adultos[2], motivo por el que comenzaron a surgir términos como glucogenosis hepática, hígado glucogénico, hepatomegalia asociada a almacenamiento de glucógeno en paciente diabético y, finalmente, Torbenson[2] en el año 2006, propuso el término hepatopatía glucogénica (HG). La mayoría de los casos descritos (62%) son mujeres, mientras que, los casos con DM tipo 2 (DM2) han sido informados con menor frecuencia (2%)[3].
La HG es una complicación que se presenta, especialmente, en individuos con DM1 mal controlada[2]. Su incidencia y prevalencia exactas se desconocen[3] debido a un descenso significativo de casos, asociado a la introducción de insulinas de acción prolongada, a la atención multidisciplinar y por considerarse, una patología infradiagnosticada y clínicamente no bien reconocida, incluso por los hepatólogos, dada su naturaleza recurrente y reversible, no completamente aclarada. La HG se caracteriza por alteraciones hepáticas como hepatomegalia y niveles elevados de transaminasas, pudiendo asociar cetoacidosis diabética (CAD)[4]. Se considera una patología benigna debido a su resolución espontánea mediante adecuado control glucémico[2].
Presentamos un caso clínico atípico de HG, por la asociación de pancreatitis de repetición y crisis epilépticas al cuadro clínico habitual. Pretendemos concienciar al clínico de esta afección infradiagnosticada, presentando un algoritmo diagnóstico que permita reducir el uso de pruebas, especialmente invasivas.
Caso Clínico
Mujer de 32 años, afecta de DM1, de 9 años de evolución, con mal control glucémico desde hace años (HbA1c 9-11%), que precisa del uso de bomba de insulina, presentando multitud de ingresos por CAD. Es valorada en el Servicio de Urgencias por cuadro clínico de dolor abdominal agudo epigástrico, con irradiación en cinturón, acompañado de náuseas, vómitos y febrícula (37,6ºC), sin semiología infecciosa concomitante.
Presenta antecedente de reiterados episodios de pancreatitis aguda alitiásica, de etiología no filiada, desde el año 2012, realizándose colecistectomía laparoscópica preventiva (julio 2014) y esfinterotomía por CPRE (colangiopancreatografía-retrógrada-endoscópica) ante sospecha de disfunción del esfínter de Oddi (diciembre 2014). Sin embargo, la paciente siguió presentando episodios de pancreatitis aguda en el contexto de CAD, destacando un episodio de pérdida de consciencia, con fijación de la mirada y movimientos tónico-clónicos siendo diagnosticada de crisis epiléptica generalizada tónico-clónica sintomática. El estudio neurorradiológico (TAC craneal y RMN cerebral) y el EEG basal fueron normales (Tabla 1).
Tabla 1
Datos de laboratorio destacados
Pruebas de imagen:
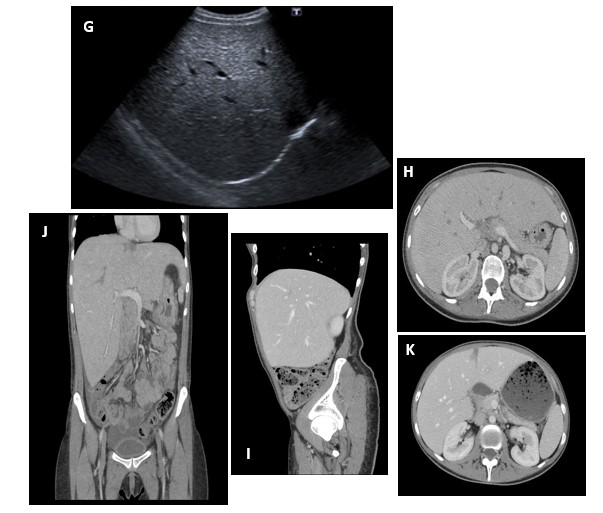
Los estudios mediante ecografía abdominal revelan hepatomegalia, parénquima discretamente hiperecogénico, de bordes regulares, sugestivo de hepatopatía difusa. El TAC toraco-abdomino-pélvico mostró hepatomegalia, con una imagen hipodensa (15 mm) en el segmento hepático IV, de características benignas, hiperdensidad homogénea del parénquima y páncreas de tamaño y captación normales. Hallazgos confirmados por la RMN de gradiente eco-dual, con desaparición posterior de la citada lesión (Figura 1).
Figura 1
Hallazgos radiológicos: Ecografía abdominal (G) Parénquima hepático con aumento de su ecogenicidad en relación con esteatosis hepática vs hepatopatía difusa. Colecistectomizada. TAC (H, I, J) Importante hepatomegalia. Hígado de alta densidad homogénea, sin lesiones focales. Páncreas con discreto engrosamiento de la cabeza de páncreas con mínima afectación de la grasa peripancreática adyacente. Moderada cantidad de líquido libre perihepático, periesplénico y en pelvis menor. Hallazgos radiológicos en el contexto clínico del paciente con pancreatitis aguda intersticial. Moderada cantidad de líquido libre en ambas gotieras parietocólicas y en pelvis menor. TAC (K) Estudio posterior con presencia de imagen hipodensa (15 mm), en el segmento hepático IV.
Estudios anatomopatológicos:
La biopsia hepática mostró una citoarquitectura normal, sin evidencia de fibrosis reticulínica significativa y ausencia de infiltrados inflamatorios, con dilatación citoplasmática hepatocitaria secundaria al acúmulo de glucógeno resultando positiva para la tinción de Schiff (PAS+) (Figura 2).
Figura 2
Hallazgos histológicos: (A, B, C) La tinción de hematoxilina y eosina (HE) muestra una arquitectura lobular portal y hepática preservada sin inflamación significativa, fibrosis o colestasis. Los hepatocitos se presentan hinchados con palidez citoplasmática y núcleos glucogenados (4x 10x 20x). (D, E, F) Fuerte positividad para la tinción ácida-Schiff (PAS) perYódica que sugiere la presencia de depósitos de glucógeno (4x 10x 20x). Los núcleos son claros y vesiculosos con frecuente pseudoinclusiones citoplasmáticas.

Discusión
Como se ha comentado, la HG es una complicación de aparición preferente en pacientes afectos de DM1 mal controlada, cuya consecuencia es la acumulación de glucógeno intrahepatocitario.
Los niveles de glucosa hepatocitarios se encuentran en equilibrio con el medio extracelular a través del transportador de glucosa GLUT-2. En presencia de altos niveles de glucosa plasmática se produce una difusión pasiva de la misma al interior celular, donde es convertida a Glucosa-6-Fosfato por la acción irreversible de la Glucoquinasa. A continuación, se transforma en Glucógeno mediante la activación alostérica de la Glucógeno Sintasa (activada por una fosfatasa estimulada por altos niveles de glucosa e insulina)[3],[4] y la inhibición de la fosforilasa glucogenolítica. Aunque los mecanismos fisiopatológicos no son bien conocidos, se cree que fluctuaciones recurrentes de los niveles de glucosa (hipoglucemia e hiperglucemia), junto con un tratamiento concomitante mediante dosis supra-fisiológicas de insulina sería el mecanismo potencial de sobreproducción de glucógeno[3],[4].
Clínicamente, la HG incluye dolor abdominal, náuseas, vómitos, y alteraciones en las pruebas de función hepática (hipertransaminasemia), cuyo origen radicaría en la eliminación celular enzimática, secundaria al acúmulo glucogénico y no a necrosis celular[2],[5]. Para interrumpir este ciclo, es necesario conseguir un periodo relativamente estable de euglucemia con el uso de insulina. Munns[6] sugirió que la administración de glucosa a pacientes con tendencia a la hipoglucemia sintomática insulínica conduciría a un mecanismo de sobreglucogenosis. Asada[5] demostró cómo la monitorización continua y mejoría del nivel máximo de glucosa postprandial ayuda a corregir la hepatopatía. Esto apoyaría el hecho de que una mínima reducción (0,6%) en la HbA1c mejora el dolor abdominal y reduce los niveles de enzimas hepáticas[7]. Por ello, una disminución de la concentración máxima de glucosa mediante riguroso control glucémico permitiría recuperar la función hepática en pocas semanas o meses, así como, la remisión completa de la clínica y las anomalías histológicas[5]. Sin embargo, no todos los pacientes deben ser diabéticos para desarrollar HG, pues, se han informado casos asociados a síndrome de dumping[8], anorexia nerviosa[9] y corticoterapia[10]. El trasplante pancreático ha llegado a producir mejoras histológicas en la entidad[11], llevando a señalar la existencia de defectos genéticos asociados con el metabolismo glucogénico como base de esta patología. Así, alteraciones de la laforina (glucógeno-fosfatasa) o mutaciones inhibitorias de la glucógeno-fosforilasa hepática[12] podrían llegar a inducir la HG, aunque, hasta la fecha, no ha sido posible identificar los genes codificantes[5].
La clínica de dolor en hemicinturón, la elevación de enzimas pancreáticas sugestiva de pancreatitis aguda, así como la aparición de crisis epilépticas generalizadas, resulta atípico. Las alteraciones pancreáticas pueden ser explicadas, por un lado, por la elevación de triglicéridos que presentan estos pacientes y, por otro, por las elevadas concentraciones de hidrogeniones asociadas a la CAD donde, la intensa vasodilatación de los pequeños vasos sanguíneos generada por esta circunstancia interferiría en la reabsorción de enzimas pancreáticas[13].
Las convulsiones como síntoma clínico de DM1 son muy raras y, respecto al mecanismo etiológico, se establece como hipótesis el descenso del umbral epileptogénico, ya que la hiperglucemia incrementa el metabolismo del ácido gamma-aminobutírico (GABA)[14]. En cambio, la cetonemia eleva sus niveles, aumentando la actividad de la decarboxilasa del ácido glutámico (GAD65), convirtiendo el glutámico en GABA, elevando el umbral epileptogénico (acción anticonvulsivante). Sin embargo, existen casos de crisis aún en presencia de cetosis, cuya explicación radicaría en la existencia de anticuerpos antiglutámico-decarboxilasa (anti-GAD), presentes en el 70-80% de pacientes DM1 y en afecciones neurológicas (síndrome Stiff-man), aunque, con títulos muy elevados respecto a DM1[14].
El retraso del crecimiento y las características cushingoides descritas clásicamente, se consideran secundarias al hipercortisolismo asociado a los periodos de hipoglucemia, siendo más frecuentes en pacientes afectos de HG.
El diagnóstico de HG descansa sobre cuatro pilares fundamentales: la clínica (descrita anteriormente), las alteraciones bioquímicas, las pruebas de imagen y la biopsia hepática.
Dentro de las alteraciones bioquímicas, destaca la hipertransaminasemia, con niveles de AST significativamente superiores a los de ALT[4], resultando excepcional el patrón colestásico. Si bien, suele mantenerse la función de síntesis hepática, es posible observar hipoalbuminemia[3]. Destacan valores de HbA1c más elevados en pacientes con DM1 y HG, que en aquellos sin HG. Esto podría contribuir a mayores niveles de triglicéridos y colesterol[3]y mayor riesgo de CAD, por alteración en la lipólisis, siendo posible que pacientes con HG presenten defectos en la biología lipídica. Un buen control glucémico mejorará las concentraciones lipídicas, disminuyendo el colesterol en 3,87 mg/dl (2,2%) y los triglicéridos en 7,1 mg/dl (8%) por cada descenso porcentual de HbA1c[15].
Por otro lado, Morton, Fitzpatrick y Brouwers[16],[17],[18] sugieren considerar los niveles de lactato plasmático secundarios a una gluconeogénesis reducida y la falta de conversión de piruvato en glucosa[17],[18], que se muestran elevados en pacientes afectos de HG, resultando potencialmente relevantes en el control evolutivo de la DM1.
Respecto a las pruebas de imagen, la ecografía abdominal muestra hepatomegalia, siendo incapaz de distinguir la HG del acúmulo graso. Morton[16] indica como la conjunción de TAC y RMN con secuencia de doble gradiente, [13]C MRS (Magnetic resonance spectroscopy) y elastografía hepática (Fibroscan®), podrían jugar un papel primordial a la hora de diferenciar ambas entidades, evitando realizar pruebas invasivas. Así, el TAC[16], muestra el hígado graso no alcohólico (HGNA) con una atenuación hepática disminuida, mientras que la HG se caracteriza por una hiperdensidad secundaria al depósito de glucógeno intrahepatocitario. No obstante, la confirmación definitiva radica en la biopsia hepática, por lo que su sensibilidad y especificidad en el diagnóstico de la HG deberían compararse en investigaciones adicionales.
La biopsia hepática guiada por ultrasonidos se considera la prueba diagnóstica "gold standard", destacando la presencia de hepatocitos hinchados y pálidos ("células fantasma") por la abundante acumulación de glucógeno intracitoplasmático. Se emplean la tinción de hematoxilina-eosina, seguida de digestión con ácido peryódico de Schiff (PAS). Histológicamente, la arquitectura se mantiene intacta, no evidenciando necrosis o colestasis, con presencia de mínima o nula inflamación, cambios grasos leves o inexistentes y ausencia de fibrosis significativa[19]. Sin embargo, si la fibrosis puede o no progresar con repetidos episodios de HG, necesita ser esclarecido.
Existen otras patologías que deben considerarse en el diagnóstico diferencial de la HG: HGNA, enfermedad de depósito de glucógeno (GSD), trastornos autoinmunes (celiaquía y hepatitis autoinmune), hepatoesclerosis, hemocromatosis, enfermedad de Wilson, hepatitis aguda por virus hepatotropos (VHA, VHB, VHC, VHD, VHE, fundamentalmente) y otros menos frecuentes del tipo VHS (Virus Herpes-Simple), VVZ (Virus Varicela-Zoster), VEB (virus Epstein-Barr) y CMV (Citomegalovirus). Dentro de los trastornos de almacenamiento del glucógeno destaca la enfermedad de Von Gierke, con hallazgos similares en la biopsia hepática. Sin embargo, la edad de presentación, la historia de DM1, el retraso en el crecimiento o la severa acidosis láctica permiten descartar su diagnóstico.
La HG, generalmente, es catalogada por los clínicos, incorrectamente, como HGNA. Sin embargo, aunque clínicamente son indistinguibles, es crucial diferenciar las dos entidades patológicas, puesto que su manejo y pronóstico son diferentes:
La HG presenta un curso benigno y funciones de síntesis hepática preservadas, asociándose principalmente con DM1 y un IMC (índice de masa corporal) bajo o normal[19]. No obstante, su recurrencia conduce a situaciones de alta morbi-mortalidad: CAD, coma hiperosmolar y/o complicaciones vasculares a largo plazo.
El HGNA es la patología hepática más común asociada con DM2 y obesidad[20], que puede causar inflamación, fibrosis, progresar a cirrosis avanzada e incluso producir hepatocarcinoma si no se instaura un tratamiento precoz[20].
Aunque la HG, generalmente, se resuelve tras un periodo de euglucemia sostenida, la esteatosis hepática puede persistir a pesar del correcto control glucémico[21]. Por lo tanto, el diagnóstico de HG conlleva información pronóstica y terapéutica importantes, y debe tenerse en cuenta en pacientes diabéticos con hepatomegalia y/o pruebas hepáticas anormales. El principal problema diagnóstico de la HG es que el examen histológico (test invasivo) es la única herramienta fiable que permite diferenciarla del HGNA[9],[11].
Conclusión
La HG es una complicación que aparece sobre todo en pacientes afectos de DM1 mal controlada, debido a la acumulación de glucógeno en los hepatocitos. Actualmente, sigue siendo una patología infradiagnosticada y, a menudo, confundida con entidades mejor conocidas en diabéticos como el HGNA. Aunque las manifestaciones clínicas son prácticamente indistinguibles, su diferenciación es muy importante por su distinto manejo y pronóstico.
La enfermedad es benigna y el estricto control de la glucemia puede proporcionar una rápida mejoría y normalización de las enzimas hepáticas.
Mediante la combinación de test bioquímicos y pruebas de imagen (no invasivas), fundamentalmente la RMN/TAC eco-dual, se puede proporcionar información importante, permitiendo ayudar al diagnóstico diferencial, excluyendo otras enfermedades hepáticas.
