
 Descargar número completo
Descargar número completo Download full issue
Download full issueCITA ESTE TRABAJO
Garrido Márquez I, Dávila Arias C, Moya Sánchez E.Tumor fibroso solitario mesentérico: claves para el diagnóstico. 2020;43(4):89-91.
Introducción
El tumor fibroso solitario (TFS) es una neoplasia infrecuente de origen mesenquimal que afecta a las partes blandas. Suelen estar muy vascularizados y son propensos a la degeneración mixoide[1]. Se pueden originar en cualquier parte del organismo, pero es más frecuente encontrarlos en el retroperitoneo y extremidades inferiores.
Su manifestación clínica más frecuente es como masa dolorosa que comprime estructuras vecinas.
Los hallazgos radiológicos son inespecíficos, por lo que en muchas ocasiones el diagnóstico definitivo se realiza mediante biopsia o extirpación quirúrgica[1],[2]. Debido a estas particularidades es importante conocer su origen, las posibles localizaciones anatómicas así como los hallazgos radiológicos más característicos que nos ayuden a identificarlo con mayor precisión.
El pronóstico generalmente es bueno. Su tratamiento definitivo consiste en extirpación quirúrgica amplia, si bien, dado que son tumores hipervascularizados, se está investigando la utilidad de los fármacos antiangiogénicos.
Caso Clínico
Presentamos el caso de una paciente de 55 años sin antecedentes personales de interés, que consultó por sensación de masa en parrilla costal izquierda, con aumento de tamaño progresivo y molestias difusas inespecíficas en epigastrio e hipocondrio izquierdo, junto con astenia y pérdida de peso de aproximadamente 5 kg en 2 meses.
A la exploración el abdomen se encontró blando y depresible con elevación en región subcostal izquierda, dura e indolora. A la palpación se apreció empastamiento y sensación de masa en hipocondrio y flanco izquierdos. Los parámetros analíticos (hemograma y bioquímica) estaban en rango de normalidad.
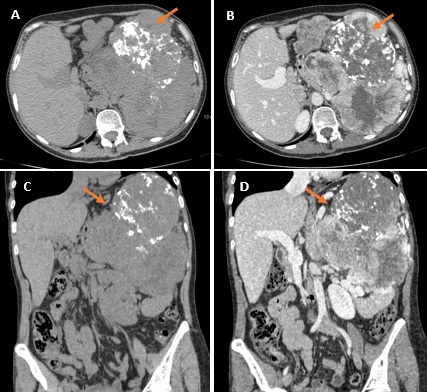
Se realizó radiografía de tórax y abdomen (Figuras 1A y 1B) en la que se apreció una masa calcificada en región de hipocondrio izquierdo por lo que se completó el estudio inicial mediante ecografía (Figuras 1B y 1C), demostrándose la presencia de dicha masa de ecogenicidad heterogénea, sin poder determinar claramente su dependencia. Para ello, se realizó tomografía computarizada (TC) abdominopélvica con contraste intravenoso en fase portal que confirmó dichos hallazgos. Se determinó la presencia de una gran tumoración sólida polilobulada de contornos bien definidos con áreas centrales de degeneración quística y calcificaciones groseras en su interior. Comprime y desplaza caudalmente al páncreas, riñón izquierdo y bazo y hacia la derecha al lóbulo hepático izquierdo, presentando íntimo contacto con el mismo (Figura 2).
Figura 1
A y B: radiografías de tórax y abdomen donde se ve una masa en hipocondrio izquierdo con calcificaciones groseras (flechas). C y D: ecografía abdominal en la que se confirma dicha masa de ecogenicidad heterogénea y con vascularización a la exploración Doppler.
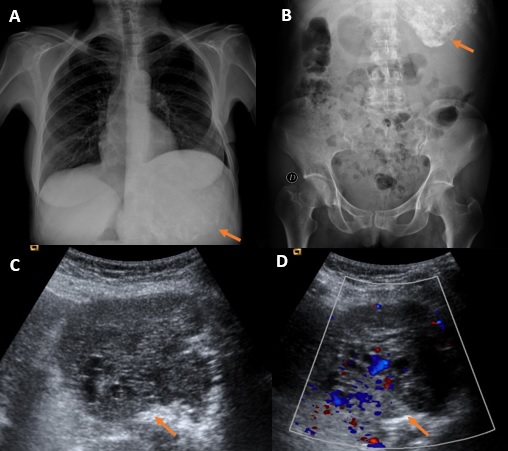
Figura 2
TC abdominopélvica axial (A y B) y reconstrucción coronal (C y D) sin y con contraste intravenoso en la que se confirman los hallazgos anteriores correspondientes a TFS (flechas): masa voluminosa de densidad heterogénea, con calcificaciones y áreas de degeneración quística, localizada en flanco izquierdo que desplaza caudalmente al páncreas, riñón izquierdo y bazo y presenta contacto íntimo con lóbulo hepático izquierdo.
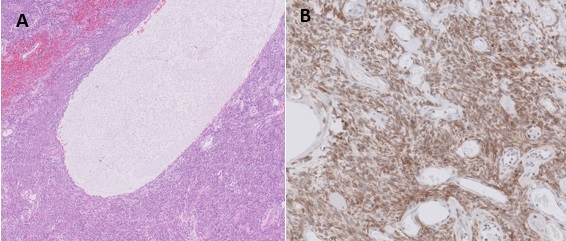
Se tomó biopsia de la tumoración con resultados histológicos e inmunohistoquímicos compatibles con TFS (Figura 3). Estos se caracterizan por tratarse de una neoplasia mesenquimal con patrón vascular ramificado. Entre las estructuras vasculares de tamaño variable se encuentran células fibroblásticas, focos de hialinización y degeneración quística con abundantes áreas de necrosis. Las técnicas de inmunohistoquímica mostraron positividad citoplásmica para CD34 y CD99 y nuclear para Bcl 2.
Figura 3
Imágenes histológicas: (A) Tinción hematoxilina eosina en la que se aprecia neoplasia mesenquimal con patrón vascular ramificado así como células fibroblásticas, focos de hialinización y degeneración quística con abundantes áreas de necrosis. (B): inmunohistoquímica que demuestra amplia positividad para el marcador nuclear Bcl-2 característico del tumor fibroso solitario.
Discusión
El TFS (conocido clásicamente como hemangiopericitoma) es una neoplasia infrecuente de origen mesenquimal que afecta a las partes blandas. Suelen estar muy vascularizados y son propensos a la degeneración mixoide.
Fue descrito por primera vez en la pleura por Klemperer y Rabin en 1931[1]. Actualmente la OMS distingue dos variantes en base a la histología: la celular, más frecuente en adultos y limitada al retroperitoneo y extremidades inferiores, y la fibrosa, que se puede encontrar en cualquier localización del organismo.
La clínica de inicio es inespecífica, siendo lo más frecuente que se manifieste como una masa poco dolorosa que comprime estructuras vecinas.
El diagnóstico definitivo se realiza mediante estudio anatomopatológico, no obstante, las técnicas de imagen ayudan al mismo. En ecografía veremos una masa relativamente definida, hipoecogénica aunque a veces su ecogenicidad es heterogénea, y con alta señal Doppler color que indica alta vascularización.
La TC con contraste intravenoso suele revelar una masa de partes blandas e hipervascularizada. Los hallazgos sugerentes de comportamiento maligno son la presencia de calcificaciones distróficas, realce heterogéneo con zonas necróticas en su interior e invasión de estructuras vecinas[2].
En resonancia magnética la masa suele ser isointensa respecto al músculo en secuencias T1 y de intensidad variable en secuencias potenciadas en T2[3]. La presencia de focos lineales o redondeados de baja intensidad indica baja celularidad, alto contenido en colágeno y bajo movimiento de protones.
Histológicamente, se aprecia una matriz colágena con células fusiformes que puede incluir áreas de necrosis, cambios quísticos o mixoides, calcificaciones, hemorragia, aumento de la vascularización, atipia o malignidad[4].
La inmunohistoquímica también juega un papel importante, encontrándose el marcador CD34 en el 75% de los tumores benignos del tórax, las proteínas Bcl-2 y vimentina en los tumores benignos de cualquier localización, y las proteínas S-100 y p53 en los malignos[5].
El pronóstico generalmente es bueno, si bien existe un pequeño porcentaje de casos (15-20%) con comportamiento maligno (6). El tratamiento definitivo consiste en extirpación quirúrgica con límites amplios, si bien se está investigando la utilidad de los fármacos antiangiogénicos dado que son tumores hipervascularizados[6].
