
 Descargar número completo
Descargar número completo Download full issue
Download full issueCORRESPONDENCIA
Eva Martínez Amate
Servicio de Aparato Digestivo
Hospital Torrecárdenas
Paraje Torrecárdenas s/n. 04009 Almería.
evamartinezamate@hotmail.com
Introducción
Presentamos el caso de un varón de 39 años diagnosticado de neoplasia pancreática a partir de pruebas de imagen que se diagnóstico finalmente de PAI a partir de otros criterios sin necesidad de confirmación histológica.
Observación clínica
Varón de 39 años que ingresó en Digestivo por cuadro epigastralgia y semiología colestásica de inicio insidioso y una semana de evolución. No refería antecedentes patológicos personales ni familiares de interés. En la exploración física presentaba ictericia cutáneo-mucosa sin otros hallazgos relevantes.
En las pruebas de laboratorio destacaba: GGT: 2095 U/L; FA: 226 U/L; BT: 2,1 mg/dl (a expensas de BD), GOT: 208 U/L; GPT: 528 U/L; amilasa: 140 U/L, con lipasa normal y sin elevación de reactantes de fase aguda. La ecografía abdominal reveló colelitiasis y aumento homogéneo y difuso del páncreas con líquido libre perivesicular y en pelvis menor, sugerente de pancreatitis aguda.
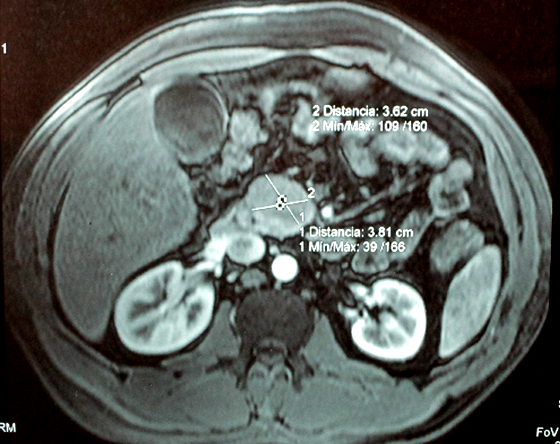
El paciente fue ingresado con el diagnóstico de posible pancreatitis aguda biliar evolucionada e ictericia de tipo obstructiva a descartar coledocolitiasis, realizándosele colangioresonancia magnética nuclear (Colangio-RMN) (Figura 1), que reveló dilatación moderada de la vía biliar intrahepática y del colédoco proximal, con marcada dilatación del Wirsung en cuerpo y cola. En cabeza de páncreas se observó LOE de 3,8 x 3,6 cm que desplazaba arteria y vena mesentérica superior, no identificándose adenopatías ni LOES hepáticas (hallazgos compatibles con neoplasia pancreática). Una ultrasonografía endoscópica (USE) y una tomografía computarizada (TAC) abdominal posteriores, no mostraron hallazgos discordantes con la Colangio-RM.
Ante estos hallazgos, se decidió realizar punción-aspiración con aguja fina (PAAF) a través de USE, no obteniéndose muestra valorable.
Posteriormente, se realizó una colangiografía retrógrada endoscópica (CPRE), observándose papila normal sin poder canular la vía biliar principal.
Durante todo el proceso, el paciente permanecía asintomático: BT: 15 mg/dl; GGT: 1364 U/l, FA: 230 U/L, siendo los marcadores tumorales normales (Ca 19,9 y CEA).
Dada la presencia de LOE pancreática y ante la imposibilidad de obtener muestra histológica a partir de las técnicas descritas, se consultó el caso con Cirugía, llegándose a la conclusión conjunta que dicha lesión focal aguda sin compromiso ganglionar y/o vascular se trataba con alta probabilidad de un tumor inflamatorio, posponiendo la resección quirúrgica de la lesión, hasta llegar a un diagnóstico preciso.
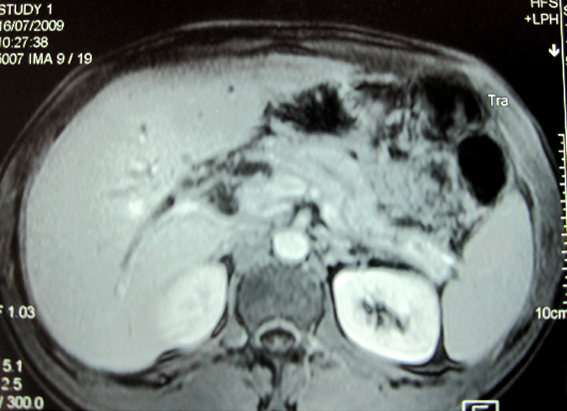
En el contexto del diagnóstico diferencial de LOEs pancreáticas, nos plantemos la posibilidad de pseudotumor pancreático como forma de expresión de PAI. Ante esta sospecha, se solicitaron inmunoglobulinas, autoanticuerpos, anticuerpos antianhidrasa carbónica I y II y antilactoferrina, encontrando la Ig G4 elevada (366 U/L) y los anticuerpos antianhidrasa carbónica I y II positivos. Basándonos en los criterios diagnósticos que recomiendan la Sociedad Japonesa de Páncreas y la Clínica Mayo, se llegó a la conclusión de alta probabilidad de PAI, por lo que se instauró tratamiento con prednisona 40 mg al día durante un mes, prosiguiéndose a pauta descendente progresivamente. En las primeras dos semanas de tratamiento, el paciente experimentó una llamativa mejoría clínica y analítica, disminuyendo la colestasis e hipertransaminasemia. Las manifestaciones clínicas, los exámenes de laboratorio y las pruebas de imagen comenzaron a mejorar a los 2 meses de iniciado el tratamiento médico, hasta llegar a normalización después de 8 meses de evolución. La respuesta al tratamiento esteroideo confirmó el diagnóstico de PAI en este paciente (Figura 2).
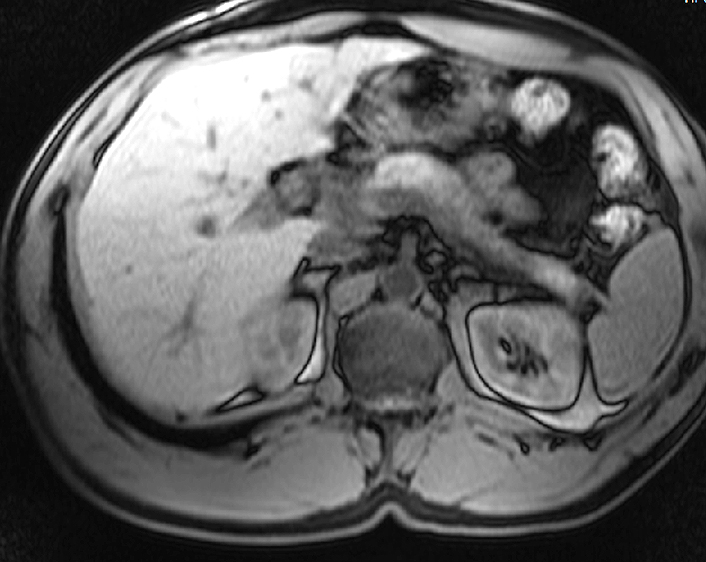
El paciente fue valorado mensualmente durante un periodo de 18 meses, donde presentó un cuadro de recurrencia clínica y analítica al disminuir la dosis de corticoides, por lo que se decidió comenzar nueva pauta de corticoides y disminuirlos más lentamente. En la actualidad, permanece asintomático y con regresión total de las pruebas de imagen (Figura 3) con dosis mínimas de corticoides.
Discusión
La PAI es una forma rara de pancreatitis crónica, de la que no existen datos aún sobre su prevalencia en países occidentales, sin embargo, sí se disponen de datos orientativos a partir de series publicada en Japón que estiman una incidencia de 0,82 diagnósticos por cada 100.000 habitantes[1]. En Occidente, los datos de incidencia de los que se disponen están basados en los diagnósticos realizados a partir de estudios histológicos sobre resecciones pancreáticas por sospecha de malignidad (2,4% al unir varias series)[2] y enfermedad benigna (11% según las series)[3].
Desde su descubrimiento en 1961[4], ha recibido múltiples denominaciones: pancreatitis esclerosante linfoplasmocitaria con colangitis, pancreatitis crónica destructiva ductal no alcohólica y pancreatitis esclerosante crónica.
Los criterios diagnósticos de esta entidad aún no están bien establecidos[5], [8]. En 2002, la Sociedad Japonesa para el estudio del Páncreas propuso unos criterios diagnósticos para la PAI (Tabla 1). Poco más tarde, Kamisawa et al.[6] sugirieron que la PAI era una enfermedad sistémica relacionada con la presencia de IgG4, para lo que se basaron en los hallazgos que tanto el páncreas, como otros órganos (vesícula, hígado, glándulas salivales, mucosa gástrica, mucosa colónica o ganglios linfáticos) presentaban: un infiltrado de células plasmáticas IgG4 positivas. Posteriormente, en 2006, estos criterios fueron revisados y dichas características se incluyeron en los criterios diagnósticos de Corea para la PAI[7]. Por último, la Clínica Mayo[8] definió la PAI como una enfermedad sistémica relacionada con la presencia de niveles elevados de Ig G4 en suero y un infiltrado de células plasmáticas IgG4 positivas con una serie de criterios clínicos, biológicos e histológicos característicos, modificados recientemente (Tabla 2).
Tabla 1
Criterios diagnósticos de PAI según la Sociedad Japonesa de Páncreas.
Tabla 2.1
Criterios diagnósticos de la PAI. Clínica Mayo.
Tabla 2.2
Grupos diagnósticos según la Clínica Mayo.
Actualmente, el concepto de PAI engloba dos entidades con características distintas: la PAI tipo 1 (pancreatitis esclerosante linfoplasmocitaria), en la que el páncreas está implicado como parte de una efermedad sistémica IgG4 positiva y, la PAI tipo 2 que se caracteriza por lesiones epiteliales pancreáticas constituídas por granulocitos, sin células IgG4 positivas y sin enfermedad sistémica asociada[8]. Aún no está claro si estos dos nuevos conceptos de PAI tienen implicaciones pronósticas diferentes y deberían ser manejados de forma distinta, aunque se ha visto que la recaída clínica después del tratamietno parece ser menos común en la PAI tipo 2. Asimismo, se ha visto que los pacientes con PAI tipo 2 son más jóvenes, tienen una baja prevalencia de elevación de la IgG4 y se asocian más frecuentemente a enfermedad inflamatoria intestinal pero menos frecuentemente a otras manifestaciones extrapancreáticas en comparación con la PAI tipo 1.
En cuanto a los criterios diagnósticos, no existe tampoco una uniformidad aceptada. Se han propuesto varios criterios, siendo los más utilizados los de la Sociedad japonesa y coreana y los de la Clínica Mayo. (Tablas 1 y 2)[9].
Desde el punto de vista clínico, los pacientes con PAI suelen presentar un cuadro de molestias abdominales de una duración variable, incluso de meses, y en ocasiones, permanecen asintomáticos. Un tercio de los pacientes presenta pérdida de peso y raramente van a desarrollar una pancreatitis aguda (PA). Otra forma de presentación es en forma de ictericia obstructiva secundaria a la estenosis del colédoco intrapancreático, que puede aparecer hasta en el 80% de los casos[10]. En muchos casos, la PAI se asocia a otras enfermedades autoinmunes (Tabla 3).
Tabla 3
Enfermedades autoinmunes asociadas a PAI.
Las pruebas de laboratorio muestran un incremento de enzimas pancreáticas, hipergammaglobulinemia, incremento de Ig G, y de forma más específica, Ig G4 junto a positividad de autoanticuerpos como anticuerpos antinucleares (ANA), anticuerpos antilactoferrina, anticuerpos antianhidrasa carbónica I y II y, con menor frecuencia, anticuerpos antimúsculo liso (ASMA) y factor reumatoide.
Los hallazgos más característicos en la TAC y RMN son el aumento de tamaño de la glándula, focal o difuso que es constante en casi todos los pacientes. En la forma difusa, la glándula aparece globulosa, homogénea, bien delimitada y de contornos lisos, con ausencia de hendiduras pancreáticas habituales, y muestra una morfología típica que se ha denominado “en salchicha”. En la forma focal, la lesión aparece como una masa que afecta con mayor frecuencia a la cabeza pancreática. Este hallazgo, junto con la presentación clínica de ictericia obstructiva, pérdida de peso y dolor abodminal insidioso, hacen que el diagnóstico diferencial entre esta entidad y el cáncer de páncreas sea difícil[11].
La histología, a pesar de ser el estándar de oro para el diagnóstico diferencial con el cáncer de páncreas, constituye uno de los criterios diagnósticos más difíciles de alcanzar, como fue en el caso de nuestro paciente.
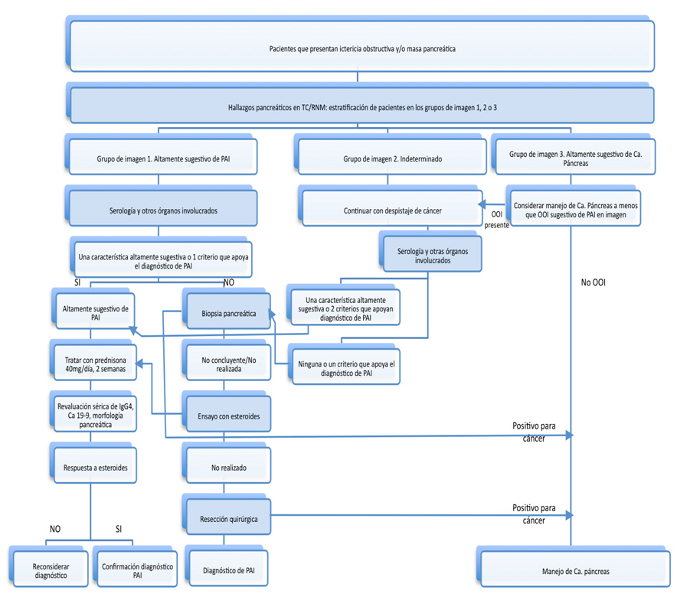
Actualmente existen dos estrategias propuestas a la hora de distinguir la PAI del cáncer de páncreas, las cuales a partir de los hallazgos de imagen estratifican a los pacientes en dos grupos: los que pueden comenzar con terapia esteroidea y los que es necesario continuar con el estudio para llegar a un diagnóstico. Una de las estrategias propuestas es la japonesa que incluye los hallazgos de la TAC y de la CPRE para estratificar a los pacientes y la otra estrategia es la americana que emplea sólo las características de la TAC para estratificar a los pacientes que necesitan una muestra histológica o no. En una reciente revisión llevada a cabo por el hospital universitario de Londres, recomiendan la estrategia americana y el manejo plasmado en la Figura 4[12].
Aunque hay publicaciones que evidencian una resolución espontánea de la PAI, los esteroides han demostrado ser efectivos en la remisión, reduciendo la frecuencia de recaídas y los efectos desfavorable a largo plazo. No obstante, aún no existen estudios controlados sobre el tratamiento de la PAI. Las series de casos publicadas con esteroides orales a distintas dosis y durante distintos períodos de tiempo demuestran unos resultados excelentes. La mayoría recomienda una dosis de 20 a 40 mg/día de prednisona y una reducción paulatina de la misma. No obstante, la frecuencia de recaída al suspender los esteroides oscila entre un 0 y un 68% de casos según las series[12]. Esto ha motivado que algunos autores propongan una terapia de mantenimiento esteroidea indefinida o bien la asociación y/o sustitución por inmunosupresores como la azatioprina[13] u otros como el micofenolato de mofetilo, el metotrexate o el rituximab[14], siguiéndose como modelo el esquema de tratamiento de una entidad cuya historia natural es similar: la hepatitis autoinmune.
Hay preguntas acerca de la PAI cuya respuesta es aún incierta, como son el valor terapéutico de estos immunosupresores, la evolución en el tiempo en relación con el tratamiento y, sobre todo, el pronóstico de la enfermedad, que en la actualidad es desconocido.
Aún son necesarios estudios a gran escala que validen los criterios diagnósticos mencionados y las distintas propuestas de tratamiento, los cuales quedan limitados por la escasa incidencia de esta patología. No obstante, la comunicación de casos como éste puede ser interesante a la hora de valorar la utilidad de estos criterios y llegar por tanto a un consenso.
