
 Descargar número completo
Descargar número completo Download full issue
Download full issueCORRESPONDENCIA
Ana Gila, Luis Rodríguez, Javier Salmerón
CIBERehd y Unidad Clínica Aparato Digestivo
Hospital Universitario San Cecilio. Granada.
anagilamedina@yahoo.es
Introducción
Aproximadamente el 3% de la población mundial está infectada con el virus de la hepatitis C (VHC), lo que representa alrededor de 175 millones de personas, con 3-4 millones de nuevas infectados cada año[1]. Alrededor del 70% desarrollan una hepatitis C crónica y aproximadamente el 20% terminan en una cirrosis hepática en un plazo de 20 años. La tasa de mortalidad después de haber desarrollado la cirrosis hepática es del 2% al 5% por año1. Actualmente la principal indicación de transplante hepático en países occidentales es la hepatopatía crónica por VHC[2].
Con el tratamiento actual, consistente en la combinación de interferón pegilado (IFNp) más ribavirina (RBV) administrado durante 12 o 72 semanas[3], se puede curar la infección por el VHC, pero las tasas de curación no son las más deseables, y esto es debido a factores relacionados tanto con el virus (carga y genotipo), como con el paciente. Además el tratamiento en algunos casos es de larga duración y conlleva efectos secundarios algunos de ellos graves. Por todo esto, es necesario buscar nuevas formas de tratamiento basadas en los fármacos ya disponibles, pero conjuntamente investigando nuevas moléculas que permitan mejorar la respuesta, acortar la duración del tratamiento y reducir los efectos secundarios.
El poder reproducir el virus en cultivos celulares[4], [5], [6] nos ha permitido conocer la organización genómica del vhc así como su ciclo vital, pudiendo desarrollar agentes que actúen inhibiendo específicamente la replicación del virus[7], [8], [9]: inhibiendo enzimas, análogos de nucleósidos frente al ARN viral e inmunomoduladores[10]. La figura 1 representa esquemáticamente el ciclo vital del vhc.
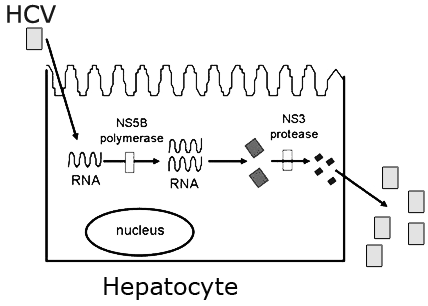
Actualmente hay dos enzimas virales, la NS3/4A proteasa y la NS5B polimerasa, que son los objetivos primordiales para el diseño de inhibidores específicos del VHC (tabla 1); varios compuestos ya han sido probados contra estas enzimas, lo cual alienta ciertas esperanzas de su uso en la práctica clínica. Otras enzimas virales, como la helicasa, la NS2 proteasa y la NS5A, hasta ahora no se han desarrollo como nuevas moléculas y sólo recientemente han sido examinados como objetivos terapéuticos. En la actualidad, estas moléculas se encuentran en diferentes fases de desarrollo y todavía no se han definido de forma adecuada la duración y las dosis óptimas de cada uno de estos agentes. Además, parece que el proceso de desarrollo será largo y que estos compuestos se añadirán inicialmente al tratamiento estándar de la hepatitis C. Uno de los problemas que se ha observado con las nuevas moléculas es la aparición de resistencias, algo similar a lo que sucede con el virus de la inmunodeficiencia humana (VIH), que parece que se podría solventar con la combinación de fármacos con mecanismos de acción diferentes. También han presentado efectos secundarios que en algunos casos han obligado a suspender la investigación.
Tabla 1
Compuestos en investigación para el tratamiento del virus de la hepatitis C (VHC).
Inhibidores de la serina proteasa
El dominio amino-terminal de la proteína NS3 del VHC y su cofactor, el NS4A, se combinan para formar una heterodímero con actividad serina proteasa, la NS3/4A, que rompe la poliproteína VHC en 4 proteínas funcionales no estructurales, incluyendo la VHC polimerasa (NS5B) (figura 2). La replicación viral se iniciará sólo después de que todas las proteínas individuales se hayan separado de la poliproteína. La proteína NS3 también ha mostrado actividad nucleótido trifosfatasa, que es una parte incorporada de la actividad helicasa. Por lo tanto, la proteína NS3 actúa como una enzima bifuncional con actividad tanto de un serina proteasa como de un ARN helicasa[12], [13].
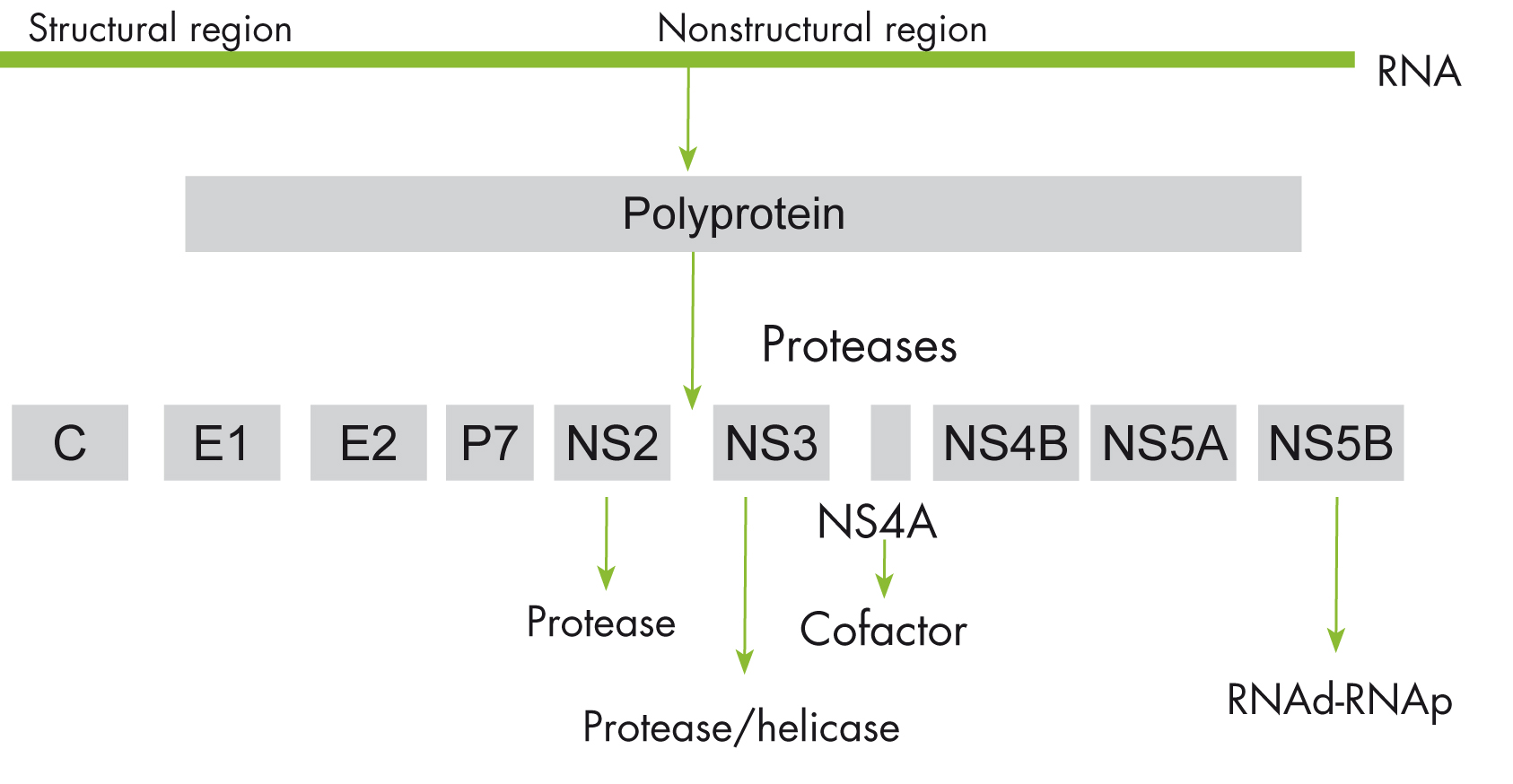
Esta serina proteasa se ha convertido en una de las dianas que más interés ha suscitado para el desarrollo de nuevas moléculas frente al VHC que, básicamente, remedan la estructura del sustrato viral sobre el que actúa la proteasa. Así, se han desarrollado dos clases de moléculas inhibidoras de proteasas. El primer grupo está representado por inhibidores no covalentes, como ciluprevir y ITMN-191/R-7227. El segundo grupo está formado por inhibidores covalentes reversibles, también conocido como inhibidores de serina-trap. Los fármacos más prometedores de esta última clase son telaprevir (TVR) y boceprevir[14] y a ellos nos vamos a referir en este apartado.
El BILN-2061 o ciluprevir, fue el primero de estos fármacos que se desarrolló y con él se consiguió reducir el ARN del VHC de modo significativo en pacientes con infección por genotipo 1 y, en menor medida, en los genotipos 2 y 3. Sin embargo, hubo de interrumpirse la investigación por toxicidad cardíaca[15].
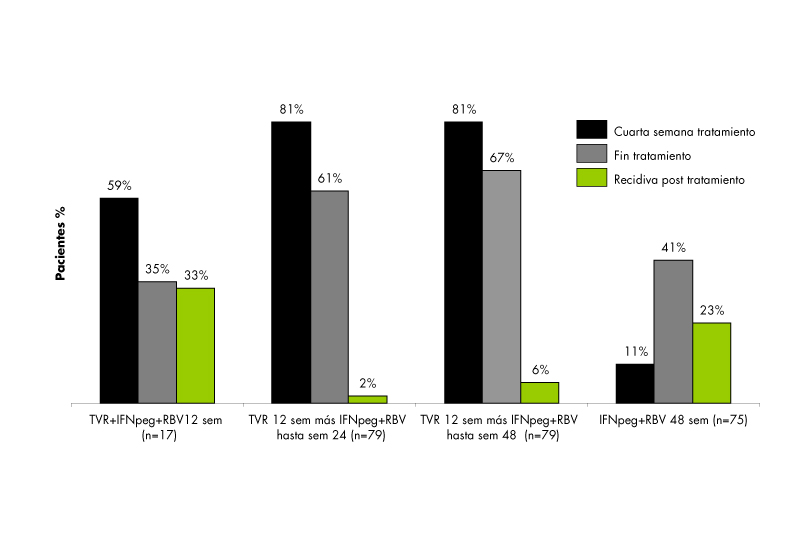
Figura 3
Resultados del ensayo PROVE-1 (análisis por intención de tratamiento)[18]. El porcentaje de pacientes con niveles de ARN sérico del virus de la hepatitis C (VHC) =10 UI/ /mL a la semana 4 (barras rojas) y el de final de tratamiento (barras verdes) y los que tienen una recidiva de los niveles de ARN después del tratamiento (barras azules) están representados para cada grupo de tratamiento. La suspensión debido a efectos adversos ocurrió en el 18% de pacientes del grupo tratado con TVR y en el 4% del grupo control. IFNpeg = IFN pegilado; RBV=ribavirina; TVR= telaprevir.
El VX-950 o telaprevir es un inhibidor peptidomimético de la proteasa NS3/4A, de administración oral que se une de forma covalente pero reversible al sitio activo de la enzima. Los principales resultados con este fármaco se han obtenido de estudios con pacientes no tratados previamente y genotipo 1[16]. En el estudio para definir la dosis más efectiva del fármaco se demostró que la administración de 750 mg cada 8 h lograba reducciones del ARN de hasta 4,4 log10 U/ml[17]. No obstante, se observó en los pacientes que seguían en tratamiento durante la segunda semana, una reactivación del ARN por selección de variantes resistentes en relación con la sustitución de serina por alanina en la posición 156. Los efectos secundarios más frecuentes fueron la cefalea y la diarrea. La combinación de telaprevir con IFN pegilado con/sin RBV evita el desarrollo de resistencias. Recientemente se han publicado datos de los ensayos clínicos fase IIb, PROVE 1 (PROtease inhibitors for Viral Evaluation) y PROVE 2 en Estados Unidos y Europa respectivamente[18], [19].
Figura 4
Resultados del ensayo clínico PROVE-2 (análisis por intención de tratamiento)19. El porcentaje de pacientes con niveles séricos de ARN del virus de la hepatitis C (VHC) = 10 IU/mL a la semana 4 (barras rojas) y del final de tratamiento (barras verdes) y de los que han experimentado recaída del nivel de ARN-VHC al final del tratamiento (barras azules) están representados para cada grupo de tratamiento. IFNpeg=IFN pegilado; RBV=ribavirina; TVR=telaprevir.
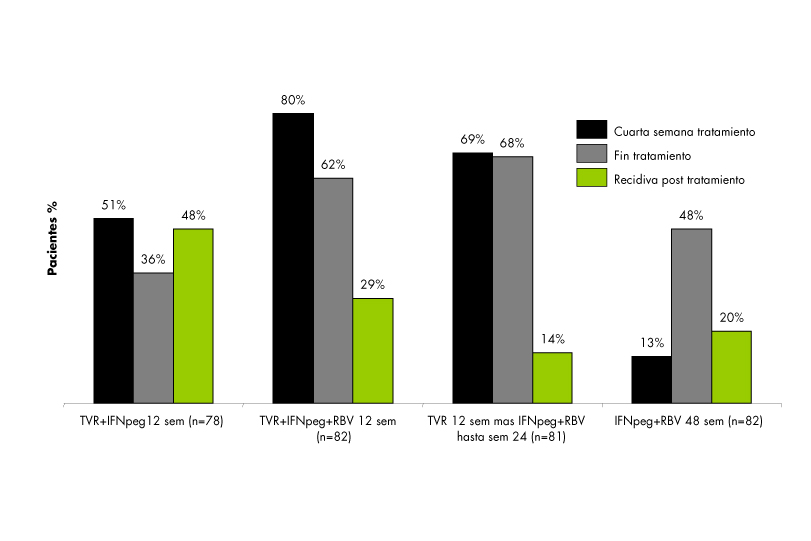
Figura 5
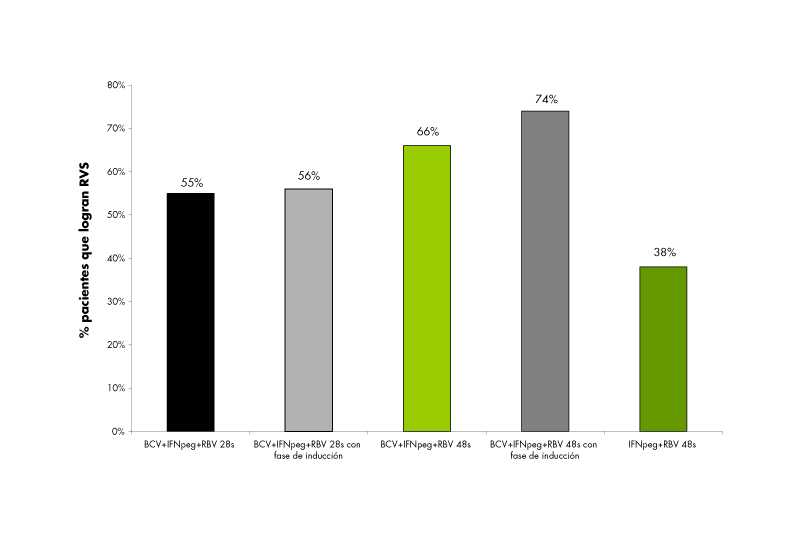
Resultados provisionales del estudio SPRINT-1 (análisis por intención de tratamiento)22. La suspensión del tratamiento por efectos adversos ocurrió entre el 9–19% de pacientes en los grupos tratados con BCV y en el 8% de pacientes en el grupo control. BCV=boceprevir; RVR=respuesta virológica rápida a las 4 semanas de tratamiento; IFNpeg=IFN pegilado; RBV=ribavirina.
En el estudio PROVE 1 (figura 3) todos los pacientes que recibieron la asociación telaprevir/IFNpeg/RBV presentaron una mayor respuesta viral rápida que el grupo control con IFNpeg y RBV (79 frente al 11%), así como una respuesta viral mayor (ARN-VHC < 10 UI/ML) a la semana 12 (70 frente a 39%). Además la tasa de respuesta virológica aumenta al prolongar el tratamiento de 24 a 48 semanas. Los efectos adversos dermatológicos y digestivos fueron más frecuentes en los grupos tratados con telaprevir. En el PROVE 2 (figura 4) se estudió la asociación de telaprevir con IFN pegilado a-2a con o sin RBV frente al tratamiento estándar en 323 pacientes naive con genotipo 1. Las tasas de respuesta viral sostenida fue mayor en los grupos tratados con telaprevir mientras que las recidivas fueron menores en los brazos que incluyeron RBV.
El TVR también se ha estudiado en combinación con IFNpeg y RBV en pacientes genotipo 1 no respondedores previamente al tratamiento estándar (ensayo PROVE-3)[20]. Recientemente se han presentado los resultados de 115 pacientes, a los que se administró triple terapia durante 12 semanas y se continuó durante 12 semanas más con IFNpeg-a 2a + RBV. En la semana 12 tras finalizar el tratamiento, el 41% de los no respondedores al tratamiento previo y el 73% de los que recidivaron tenían el ARN del VHC indetectable. En pacientes no respondedores a tratamientos previos también se ha evaluado la triple terapia (telaprevir 750 mg/8 h + IFNpeg-a2a + ribavirina) durante 12 semanas seguida de IFNpeg-a2a + ribavirina. Aunque el número de pacientes que se han evaluado es muy pequeño, los resultados demuestran que casi todos los pacientes negativizan el ARN en la semana 12 con esta estrategia terapéutica[21].
Figura 6
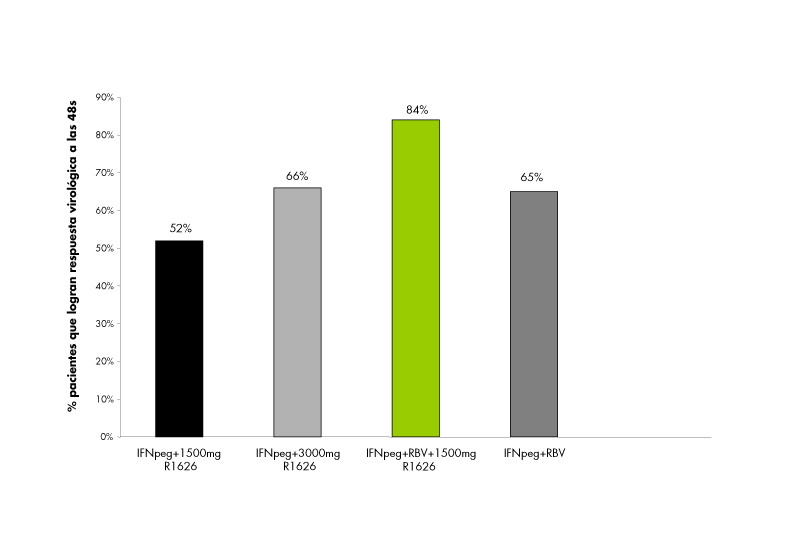
Respuesta al final de tratamiento con R1626. ARN-VHC indetectable a las 48 semanas de tratamiento.
El SCH-503064 o boceprevir es otro fármaco inhibidor de la NS3 proteasa del VHC que ha demostrado ser seguro y con buena tolerancia cuando se ha asociado al IFNpeg. Es menos potente que el telaprevir, pero también ha demostrado tener menor toxicidad cutánea. Un estudio reciente fase III, el SPRINT-1,[22] ha arrojado datos preliminares muy interesantes. Se ha realizado en 520 pacientes infectados por genotipo 1 y no tratados previamente, que se distribuyeron aleatoriamente en varios grupos. En dos de ellos se administró IFNpeg-a2b + RBV durante 4 semanas y después se añadió boceprevir (800 mg/8 h) durante 24 o 44 semanas. En otros 2 grupos se administró IFNpeg-a2b + RBV + boceprevir durante 28 o 48 semanas. Y en el grupo control se administró el tratamiento estándar (IFNpeg-a2b + RBV) durante 48 semanas. Los resultados disponibles son de la semana 12 tras finalizar el tratamiento en los grupos de 48 semanas de duración y de la semana 24 en los de 28 semanas. La tasa de respuesta mantenida en los 2 grupos tratados durante 28 semanas fue del 55 y el 56%. En el grupo tratado con triple tratamiento desde el comienzo, la tasa de respuesta mantenida fue del 66%, mientras que en el que fue tratado inicialmente con IFNpeg-a2b + RBV y se añadió boceprevir en la semana 4 y hasta la semana 48, la tasa de respuesta mantenida fue del 74% (figura 5).
Actualmente está en marcha el estudio SPRINT-2, con más de 1.000 pacientes y 3 grupos de tratamiento. Durante 4 semanas se administra a todos IFNpeg-a2b + RBV. Posteriormente se continúa hasta la semana 48 con IFNpeg-a2b + RBV + boceprevir/placebo en 2 de los grupos. En el tercero se administra el triple tratamiento hasta la semana 28 en los pacientes en los que el ARN es negativo en la semana 8, y en los que tienen ARN positivo en la semana 8 se administra el triple tratamiento hasta la semana 28 y a partir de entonces se utiliza IFNpeg-a2b + RBV + placebo.
Inhibidores de la arn polimerasa
La proteína NS5B es la principal enzima responsable de la replicación viral y por ello constituye otra diana terapéutica debido a su actividad ARN polimerasa depediente de ARN (RdRp) (figura1). Se han desarrollado dos tipos de inhibidores de la ARN polimerasa: a) los análogos nucleósidos que inhiben la iniciación o elongación de la cadena de ARN, y b) los análogos no nucleósidos que bloquean la iniciación de la polimerización del ARN-VHC (tabla 1). La mayoría de estos fármacos se encuentran en fases I y II.
Valopicitabina es un fármaco que, asociado con IFNpeg-a2a, presentó resultados esperanzadores en pacientes infectados por el genotipo 1, tanto näive como no respondedores a tratamientos previos, ya que induce un descenso del ARN-VHC dependiente de la dosis. Sin embargo, el desarrollo de este fármaco se suspendió por la mala tolerancia y los efectos adversos gastrointestinales[23].
Balapiravir o R1626 es el profármaco oral de un potente análogo de nucleósido inhibidor de la polimerasa (R1479), que es activo frente a todos los genotipos y con una barrera alta a las resistencias virales. La reducción de ARN es dependiente de la dosis, alcanzándose descensos de hasta 3,7 log10 U/ml tras 14 días de monoterapia en pacientes infectados por genotipo 1 y con dosis de 1.500-4.500 mg/día[24]. La combinación con IFNpeg-a2a y RBV tiene un efecto sinérgico, como se ha demostrado en un ensayo fase 2B en el que se incluyó a pacientes näive infectados por genotipo 1[25]. Se observó que al finalizar el tratamiento, el 84% de los pacientes que recibieron triple terapia logró la negativización del ARN frente al 65% de los tratados con el régimen estandar (figura 6). El efecto adverso más frecuente, pero reversible, fue la toxicidad hematológica (neutropenia), especialmente con las dosis más altas de balapiravir, y no se desarrollaron resistencias al fármaco.
R7128 es otro análogo de nucleósido inhibidor de la polimerasa de administración oral y que tiene efectos aditivos cuando se añade al tratamiento estándar. Se han realizado estudios tanto en pacientes con genotipo [26] y con genotipos 2/3 y no respondedores[27], obteniéndose buenos resultados preliminares. Entre los inhibidores no análogos de nucleósidos se han desarrollado varias moléculas, aunque la mayoría de ellas no han pasado a fases más avanzadas de la investigación por falta de eficacia, o por efectos secundarios o por la aparición de resistencias.
Inhibidores de la ciclofilina
La ciclofilina B participa en la replicación viral del VHC uniéndose y estimulando a la ARN polimerasa16. Se ha observado que los inhibidores de la ciclofilina, como la ciclosporina A, tienen actividad frente al VHC, pero su efecto inmunodepresor impide su desarrollo como antiviral. Así pues se han desarrollado derivados de la ciclosporina sin el efecto inmunodepresor para el tratamiento de la infección por VHC.
DEBIO-025 es uno de estos fármacos que han demostrado reducciones significativas de la carga viral cuando se asocia IFNpeg e impide la unión NS5B-ARN-VHC y la polimerización del ARN-VHC al inhibir la ciclofilina B28.
En resumen, el futuro del tratamiento de la hepatitis crónica C incluye el IFNpeg, la RBV y alguno de los nuevos antivirales usados en combinación y probablemente durante periodos de tiempo más cortos que el del régimen estándar. Todo ello con el objetivo de evitar períodos de inhibición subóptima de la replicación viral que permitieran el desarrollo de cepas resistentes[16]. Una de las combinaciones que resulta más atractiva es la de un inhibidor de la polimerasa con uno de la proteasa. Estudios realizados in vitro con fármacos que todavía no se han analizado adecuadamente en pacientes, concluyen que la combinación de fármacos consigue disminuir de manera significativa la tasa de resistencias.
